Strategidokument
Mistænkes hos personer med muskelsymptomer samtidig med anden organinvolvering. Udredning skal ske på højtspecialiseret afdeling (Neuromuskulær klinik).
Opstår på baggrund af mutation i nDNA (nukleært DNA) (Mendelsk nedarving) eller mutation I mtDNA (mitokondrie DNA) (maternel nedarving).
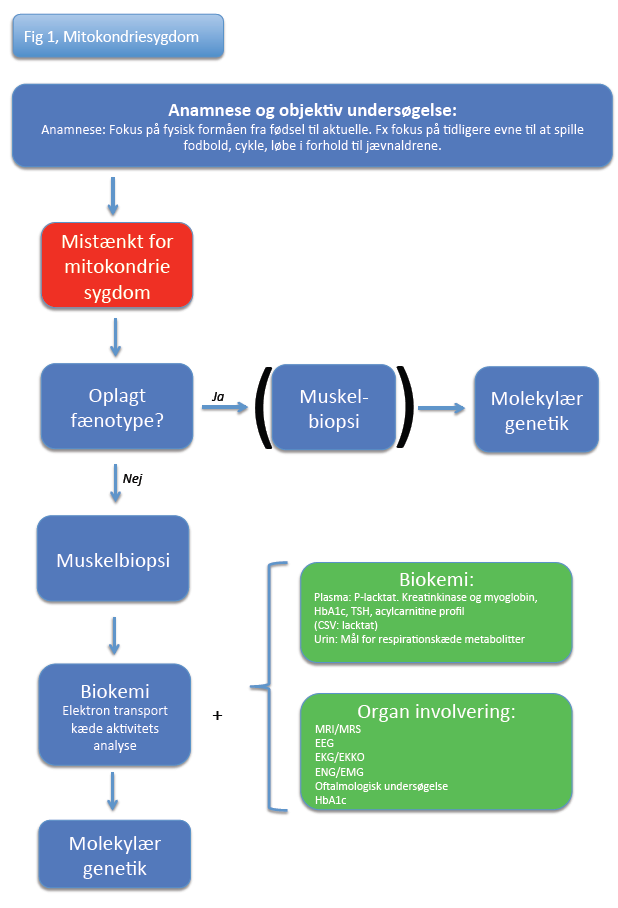
Diagnostisk algoritme, Figur 1:
nDNA og mtDNA
mtDNA indeholder kun 1% af de 3000 gener som er nødvendige for at mitokondrier bliver dannet og kan varetage de vævsspecifikke opgaver som de har udover at danne ATP til cellen. Ca. 2970 gener udtrykkes af nDNA.
En mutation i en af disse gener kan derfor medføre defekt replikation, defekt ATP produktion eller defekt i den vævsspecifikke opgave som mitokondriet varetager (som eksempel er mitokondriet det hastighedsbegrænsende trin i pyrimidinmetabolismen, som bruges til at danne RNA og DNA, mitokondrierne står for syntesen af hæm, som bruges til at danne hæmoglobin osv).
Mitokondriesygdom på baggrund af mutation i mtDNA skyldes:
- Store sporadiske rearrangementer der resulterer i deletioner og duplikationer
- Punkt mutationer eller
- Maternelt nedarvede rearrangementer (duplikationer).
Mutationer kan opstå sporadisk i germinal vævet (maternel nedarving, hyppigst punkt mutation) eller i somatiske celler (sporadiske mutationer, hyppigst large-scale deletioner).
Det humane mtDNA er dobbelt strenget, indeholder 16.569 basepar. Det repliceres og transkriberes i det mitokondrielle matrix. Hvert mitokondrie indeholder 2-10 kopier mtDNA. mtDNA koder for 37 gener: 22 tRNA; 2 rRNA til protein syntese, 13 koder for subunits til respirationskædens komplekser (7 subunits til kompleks I, 1 subunits I kompleks III, 3 subunits til kompleks IV og 2 subunits til kompleks V).
nDNA koder for øvrige subunits i respirationskædens komplekser, samt for faktorer der styrer mtDNA replikation, transkription og translokation. Mutationer i nDNA gener der koder for disse faktorer vil derfor også lede til mitokondriesygdomme og give lignende symptomer som mutationer i mtDNA.
Heteroplasmi
mtDNA repliceres ikke på samme tid som selve cellen; der sker derfor en ulig fordeling af antallet af mtDNA blandt celler, både mellem væv og imellem celler i det samme væv. På denne måde fordeles muteret mtDNA også ulig mellem alle celler inkl. ægceller. Ægceller har derfor forskellig mutationsbyrde. Søskende af moder, der har mutation i mtDNA, har derfor ofte forskellig mutationsbyrde. Ulig fordeling af muteret antal mtDNA kopier resulterer også i stor heterogenicitet både mellem patienter, mellem væv hos den enkelte patient og selv mellem celler i det samme væv.
Phenotype
Det specielle ved mitokondriesygdom forårsaget af mutation i mtDNA er at der både er muterede og wild-type mtDNA i den samme celle. Jo højere mutationsgrad (muterede mtDNA/wild type mtDNA) desto højere sandsynlighed for at de resterende wild-type mtDNA ikke kan levere den fornødne energi til cellen. Hvert væv har en grænse for, hvor meget muteret mtDNA den kan klare, før der kommer symptomer fra vævet.
Mutationsgrad over tid
mtDNA mutationsgraden kan ændre sig over tid i de forskellige væv, hvor der kan ske både en selektion for og imod muterede mtDNA kopier afhængig af det enkelte væv og genotypen.
Organinvolvering, Figur 2:
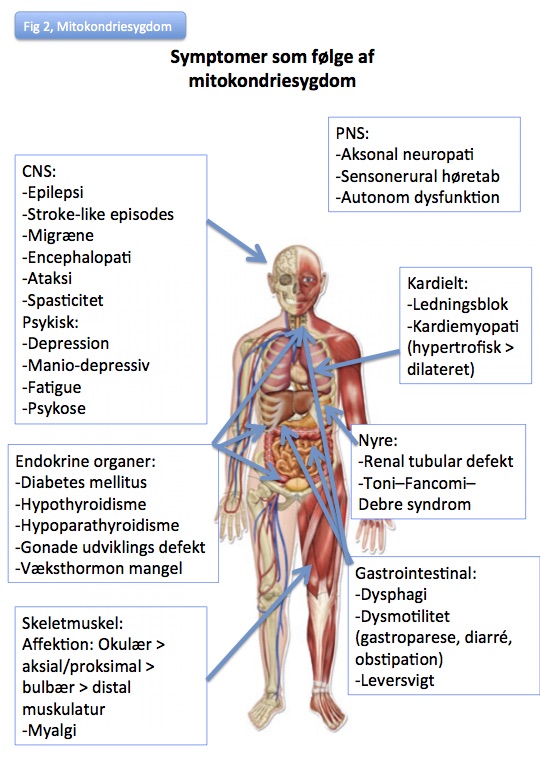
Mitokondrier leverer energi til cellen, hvorfor symptomer opstår fra celler med højt energikrav:
Nervesystem (PNS, CNS, autonome nervesystem, n. opticus og retinae)
Muskel (skelemuskel, øjenmuskel (særligt etraokulære), hjertemuskel)
Endokrine organer
Skeletmuskel
Arbejdsintolerance er det mest forekommende symptom hos patienter med mitokondriesygdom. Det skyldes at skelet muskel er det eneste organ der kan øge sin energi omsætning med en faktor 50-100 på få sekunder (fra hvile til løb).
Syn
Øjenmuskel:
CPEO (chronic progressive external ophtalmopleia) er særligt associeret med patienter med single large-scale deletion men kan også ses hos patienter med punkt mutation i mtDNA og nDNA.
Retina:
Mitokondrie sygdom er ofte associeret med pigmenteringsdefekt i retina.
N. opticus:
Særligt associeret med Lebers hereditære optisk neuropati.
Hjerte
Kardiomyopati (oftest hypertrofi af ventre ventrikel) og ledningsblokd (AV blok, grad I-III). Sidstnævnte ses særligt hos patienter med single large-scale deletion med Kearns Sayre phenotype.
Lever
Kun hos børn med mitokondriesygdomme. Isoleret forhøjet ALAT og LDH hos voksne er ikke tegn på leverpåvirkning, men udtryk for pågående muskelnedbrydning.
Endokrine organer
Beta-celler, pancreas:
Beta celler i pancreas er sidestillet med ikke-mitotisk væv, og derfor ofte afficeret hos patienter med mutation in mDNA. Man mener at 1-2% af alle patienter med DMII har dette på baggrund af mitokondrie sygdom.
Vækst hormon:
I barnealderen kan ses et lavt niveau af væksthormon hos patienter med punkt mutation i mtDNA.
PNS
Perifer neuropati, sensorisk-motorisk aksonal degenerativ neuropati.
CNS
Encephalopati:
Er et hyppigt forekommende symptom hos patienter med højt niveau af mtDNA mutationer, men er også associeret med specifikke syndromer; således er Leighs syndrom (kan skyldes både mutation i nDNA, hyppigst SURF-1 gene og mtDNA, hyppigst mtDNA 8993 punkt mutation) associeret med bilateral subakut nekrotiserende encephalopati med karakteristiske fund af symmetrisk bilaterale fund oftest i putamen, thalamus, substantiia nigra, nucleus dentatus, og periventrikulær hvid substans.
Epilepsi:
Visse phenotyper (MERFF, ragged red fibers og myoklon epilepsy) er beskrevet med epilepsi, men alle patienter med et højt niveau af mtDNA mutation kan udvikle epilepsi. Man mener at 1-2% af alle patienter med epilepsi har dette på baggrund af mitokondriedysfunktion. Epilepsien hos disse patienter kan være svær at behandle til anfaldsfrihed.
Stroke-like episodes:
Skal mistænkes hos patienter <40 år med stroke-lignende symptomer, hvor man ved CT-angio ikke finder stenose af kar. Årsag til symptomer skyldes ophobning af laktat i afficeret område.
Diagnostisk algoritme som PDF: fig-1-mm
Organinvolvering som PDF: fig-2-mm
Senest revideret d. 05.05.2022
Forfattere: Tina Dysgaard
Referenter: John Vissing, Nanna Witting og Christian Krarup
Godkender: Tina Dysgaard, redaktionsgruppe G
Keywords: mtDNA, heteroplasmi, encephalopati, stroke-like, epilepsi