Strategidokument
Formål:
Dokumentets formål er at give et indblik i den aktuelle viden om genetiske aspekter af ALS, dels som baggrund for information til ALS patienter og dels med henblik på at vurdere, om patienten bør
henvises til yderligere genetisk rådgivning
Forkortelser:
ALS – amyotrofisk lateral sclerose
FALS – familiær amyotrofisk lateral sclerose
SALS – sporadisk amyotrofisk lateral sclerose
MND – motorneuron sygdom
NGC – Nationalt Genom Center
Baggrund:
Ved ALS er de underliggende patofysiologiske mekanismer oftest multifaktorielle og ukendte. De fleste tilfælde forekommer sporadisk, men hos en undergruppe af patienterne forekommer sygdommen familiært. I dag kendes gener, som er associeret med både familiær ALS (FALS) og sporadisk ALS (SALS) (figur 1).
Arvegangen er i de monogent betingede tilfælde hyppigst autosomal dominant og langt sjældnere autosomal recessiv.
Det anslås at ca. 10 % af ALS tilfælde er familiære/monogent betingede. Tilsyneladende sporadiske tilfælde kan imidlertid have monogen ætiologi, da der findes patogene varianter, som er karakteriseret ved nedsat penetrans eller aldersafhængig penetrans. Desuden kan samme patogene variant i samme familie forårsage meget forskellige kliniske fænotyper med enten ALS, frontotemporal demens (FTD) eller parkinsonisme (se afsnittet om C9orf72), og det har derfor ikke
nødvendigvis været klart i familien, at det var variationer af samme sygdom. Der kan være tale om tabuisering/fornægtelse af sygdommen, hvilket ligesom små familier, tidlig død af anden årsag eller tidlig symptomdebut hos et barn af et endnu ikke afficeret familiemedlem, kan medføre, at familieanamnesen ikke umiddelbart afslører, at der er tale om FALS (1).
Gener:
De to hyppigste gener involveret i ALS:
- C9orf72 er det gen, der hyppigst er associeret til både FALS og SALS. Arvegangen er autosomal dominant. Den patogene variant er en hexanucleotid-repeat forlængelse, og der er ikke beskrevet andre patogene varianter i dette gen. Hexanucleotidforlængelsen findes hos ca. 40 % med FALS og hos ca. 10 % med SALS. Hvis fænotypen i familien er ALS hos nogle, FTD hos andre eller en fænotype med blandet ALS og FTD påvises hexanucleotidforlængelsen hos helt op mod 80%. Fænotypen kan også være med parkinsonisme. Penetransen vurderes at være høj, men ikke 100 %. Middel-debutalderen er 58 år, hvilket ikke adskiller sig fra debutalderen hos ALS patienter generelt. Variationen er dog betydelig, og der er beskrevet symptomdebut fra 27 til 74 år(2).
- I SOD1-genet er der beskrevet mange forskellige patogene varianter. Arvegangen er hyppigst autosomal dominant, men en hyppig missense-mutation i den skandinaviske befolkning, p.Asp90Ala, medfører en autosomal recessiv arvelig form, hvor progressions-hastigheden er langsommere. SOD1-mutationer identificeres hos 15-20 % med FALS, og hos 2-7 % med SALS. Fænotypen er sjældent med kognitiv påvirkning. Penetransen vurderes at være høj, men ikke 100
%. Debutalderen er gennemsnitligt 55 år, hvilket kun er marginalt tidligere end ALS patienter generelt. Der er dog beskrevet debut fra 13 til 79 år (2).
Patogene varianter i de øvrige gener er sjældnere og omtales ikke i detaljer her.
Information/rådgivning:
Grundig afdækning af familiær disposition til både ALS, demens og andre typer af neurodegenerative sygdomme afgør, hvorvidt patientens sygdom opfattes som SALS eller FALS.
- Hvis der er en kendt patogen variant i familien, og patienten viser tegn på ALS, kan der foretages molekylærgenetisk undersøgelse, hvis patienten ønsker det, med henblik på at sikre diagnosen tidligere, men den genetiske test indgår ikke i de diagnostiske kriterier.
- Hvis der er mistanke om FALS, kan patienten tilbydes molekylærgenetisk undersøgelse. Der bør informeres om, hvilke implikationer beslutningen kan have for den øvrige familie. Det er
patienten selv, der afgør, om han/hun ønsker undersøgelsen, men evt. modsatrettede ønsker i familien bør adresseres. Patienten og/eller andre familiemedlemmer kan henvises til genetisk rådgivning. - Hvis patienten har SALS, bør patienten informeres om, at man ikke sikkert kan udelukke, at der er tale om en arvelig tilstand, idet 10 % alligevel viser sig at have monogen ætiologi. I disse tilfælde er arvegangen autosomal dominant. Man kan ikke udelukke arvelighed ved testning, og ved tvivlstilfælde, hvilket er hyppigt i denne situation, bør patienten henvises til genetisk rådgivning.
Molekylærgenetisk undersøgelse: - MND er blandt indikationerne for helgenomsekventering i NGC og patienterne kan derfor tilbydes genetisk undersøgelse i dette regi efter visse kriterier som ses i NGC-vejledningen neurogenetiske patienter indikationer og kriterier for rekvirering af helgenomsekventering: https://ngc.dk/Media/637932988313910818/Neurogenetiske%20patienter%20-%20indikationer%20og%20kriterier.pdf
- Forudgående skal der være undersøgt for repeatekspansion i C9ORF72.
- Der kan i øvrigt testes for patogene varianter i mange gener, herunder C9orf72, SOD1, FUS, TARDBP, VCP, CHMP2B, UBQLN2, TBK1, OPTN og SETX i Molekylærgenetisk Laboratorium, Rigshospitalet: https://labportal.rh.dk/LabPortal.asp?Mode=View&Id=7086
- Det påhviler klinikeren, at der forinden skal indhentes informeret samtykke til Nationalt Genom Center: https://ngc.dk/blanketter-og-vejledninger
- Patienten bør være informeret om, at man i en del tilfælde finder ændringer, hvor man ikke med sikkerhed kan sige, om de er sygdomsfremkaldende eller ej.
- Påvisning af en patogen variant har betydning for mulighederne for at kunne tilbyde prædiktiv gentest i familien, og for mulighederne for prænatal diagnostik og præimplantationsdiagnostik ligesom det i visse tilfælde kan være muligt at tilbyde forskningsbehandling. Høj genetisk risiko for frontotemporal demens har betydning for prognosen og er kontraindikation til invasiv hjemmerespiratorbehandling.
Vedrørende muligheder for prædiktiv gentest for en kendt patogen variant i en familie henvises til afsnittet om prædiktiv gentest: http://neuro.dk/wordpress/nnbv/praesymptomatisk-genetisk-radgivning/
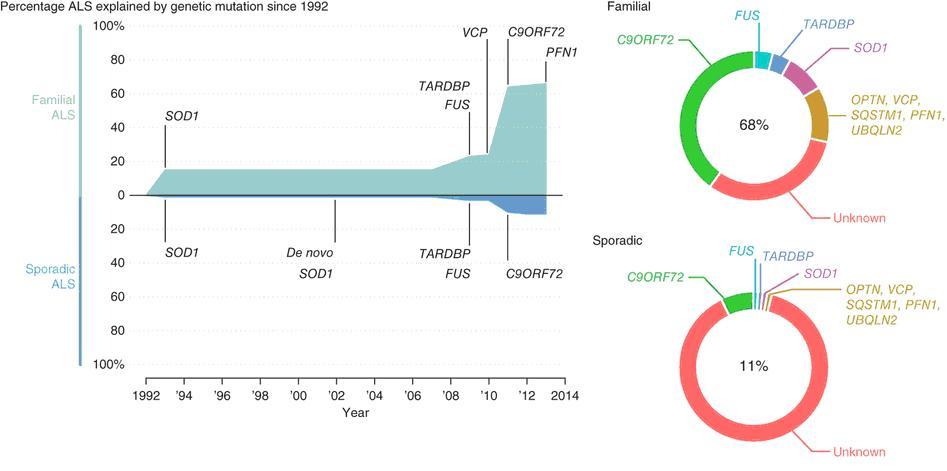
Figur 1: Gener ved FALS og SALS (1), from Renton AE, Chiò A &Traynor BJ, Nature Neuroscience17, 17–23(2014)
Referencer:
- Lindquist SG, Dunø M, Svenstrup K, Nielsen JE. Genetisk rådgivning er relevant både ved familiære og sporadiske tilfælde af amyotrofisk lateral sklerose. Ugeskr Laeger. 2014 Oct 20;176(43)
- ALS Online Database http://alsod.iop.kcl.ac.uk/
Senest revideret d. 15.02.2023
Forfattere: Kirsten Svenstrup og Suzanne Lindquist
Godkender: Nanna Winther Dombernowsky, redaktionsgruppe D